Consultant: I can help find a liver donor for your transplant surgery. From my donating clients only 4.8% (3 out of 62) had complications which is way below the national average of 10%!
Question:Is this evidence that the consultant’s work meaningfully contributes to reducing complications?
Correlation vs. causation
Epistemologic question:Is it possible to assess the consultant’s claim using the data?
No. The causal claim we can not analyze, the data is observational. Reasons can be outside of the numbers: For example, patients who can afford a medical consultant can afford better medical care, which could lead to a lower complication rate.
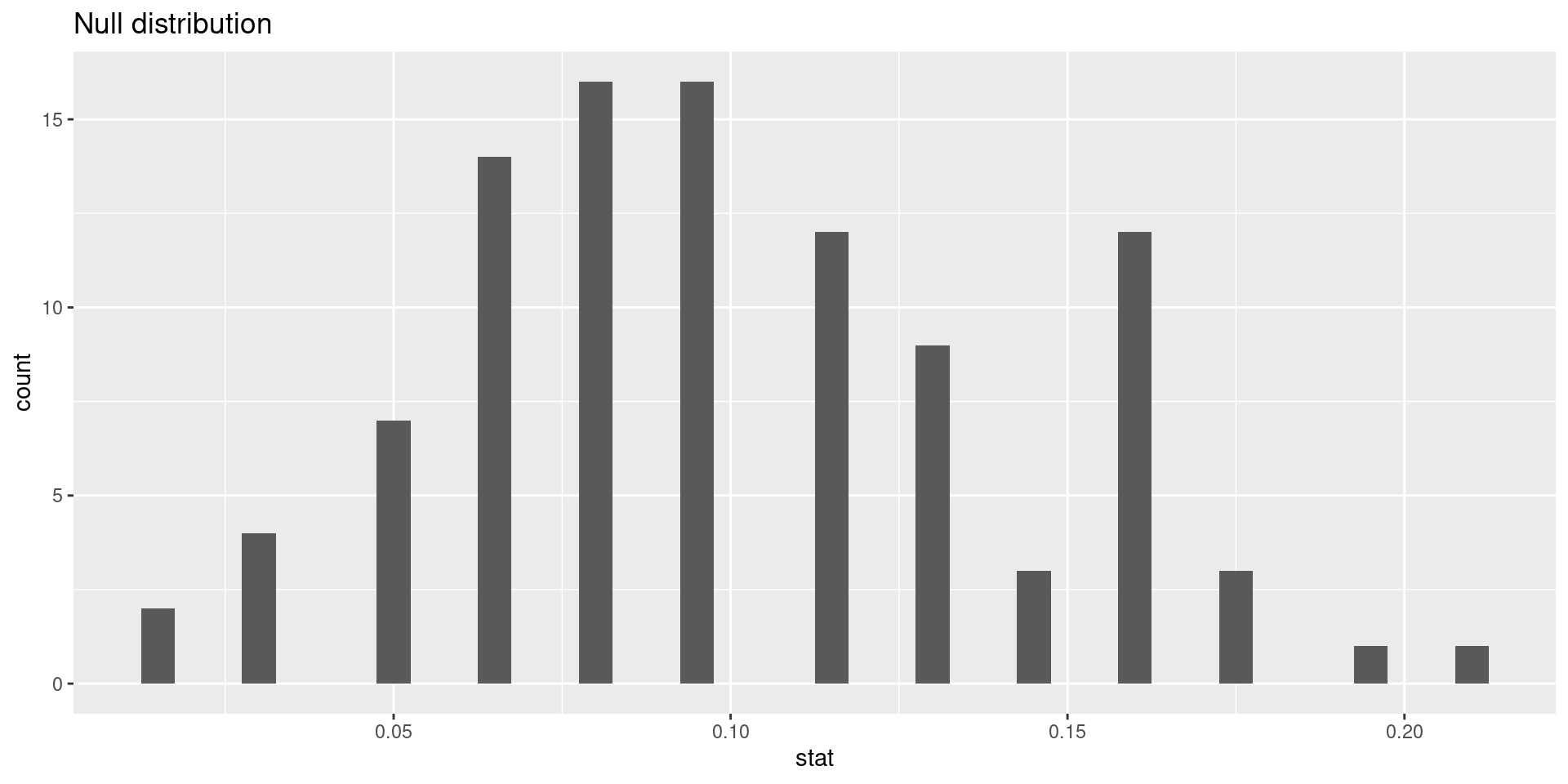
Statistics Question:Could the low complication rate of 4.8% be due to chance?
# A tibble: 62 × 1
outcome
<chr>
1 complication
2 complication
3 complication
4 no complication
5 no complication
6 no complication
7 no complication
8 no complication
9 no complication
10 no complication
# ℹ 52 more rows
organ_donor |>count(outcome)
# A tibble: 2 × 2
outcome n
<chr> <int>
1 complication 3
2 no complication 59
In a test, a parameter is the “true” value of interest.
We typically estimate the parameter using a sample statistic as a point estimate.
\(p\): true rate of complication, here 10%
\(\hat{p}\): rate of complication in the sample = \(\frac{3}{62}\) = 0.048
Two claims:
Null hypothesis \(H_0\):There is nothing going on
Complication rate for this consultant is no different than the US average of 10%
Alternative hypothesis \(H_A\):There is something going on
Complication rate for this consultant is lower than the US average of 10%
Hypothesis testing as a court trial
Null hypothesis, \(H_0\): Defendant is innocent
Alternative hypothesis, \(H_A\): Defendant is guilty
Present the evidence: Collect data
Judge the evidence: “Could these data plausibly have happened by chance if the null hypothesis were true?”
Yes: Fail to reject \(H_0\)
No: Reject \(H_0\)
Hypothesis testing framework
Start with a null hypothesis, \(H_0\), that represents the status quo
Set an alternative hypothesis, \(H_A\), that represents the research question, i.e. what we are testing for
Conduct a hypothesis test under the assumption that the null hypothesis is true and calculate a p-value. Definition:Probability of the observed outcome or a more extreme one given that the null hypothesis is true.
if the test results suggest that the data do not provide convincing evidence for the alternative hypothesis, stick with the null hypothesis
if they do, then reject the null hypothesis in favor of the alternative
Setting the hypotheses
In the following \(p\) is the “true” rate of complication of the consultant.
\(H_0: p = 0.10\) In the long run the consultant would also have a 10% complication rate.
\(H_A: p < 0.10\) Also in the long run the consultant would have a complication rate less than 10%. (We would still not know if it is because of the consultant’s work or for something else!)
Simulating the null distribution
Since \(H_0: p = 0.10\), we need to simulate a null distribution where the probability of success (complication) for each trial (patient) is 0.10.
How should we simulate the null distribution for this study using a bag of chips?
How many chips? For example 10 which makes 10% choices possible
How many colors? 2
What should colors represent? “complication”, “no complication”
How many draws? 62 as the data
With replacement or without replacement? With replacement
When sampling from the null distribution, what would be the expected proportion of “complications”? 0.1
[1] "no complication" "no complication" "no complication" "no complication"
[5] "no complication" "no complication" "no complication" "no complication"
[9] "no complication" "no complication" "no complication" "no complication"
[13] "no complication" "complication" "no complication" "no complication"
[17] "no complication" "no complication" "no complication" "no complication"
[21] "no complication" "no complication" "no complication" "no complication"
[25] "no complication" "no complication" "no complication" "complication"
[29] "no complication" "no complication" "no complication" "no complication"
[33] "no complication" "no complication" "no complication" "no complication"
[37] "no complication" "no complication" "complication" "no complication"
[41] "no complication" "no complication" "no complication" "no complication"
[45] "no complication" "no complication" "no complication" "no complication"
[49] "no complication" "no complication" "no complication" "no complication"
[53] "no complication" "no complication" "no complication" "no complication"
[57] "no complication" "no complication" "no complication" "no complication"
[61] "no complication" "no complication"
sum(sim1 =="complication")/62
[1] 0.0483871
Oh OK, this is the same as the consultant’s rate. But maybe it was a rare event?
Oh OK, our fist simulation with 0.0483871 seems to be comparably low.
Automating with tidymodels1
organ_donor
# A tibble: 62 × 1
outcome
<chr>
1 complication
2 complication
3 complication
4 no complication
5 no complication
6 no complication
7 no complication
8 no complication
9 no complication
10 no complication
# ℹ 52 more rows
library(tidymodels)set.seed(10)null_dist <- organ_donor |>specify(response = outcome, success ="complication") |>hypothesize(null ="point", p =c("complication"=0.10, "no complication"=0.90)) |>generate(reps =100, type ="draw") |>calculate(stat ="prop")null_dist
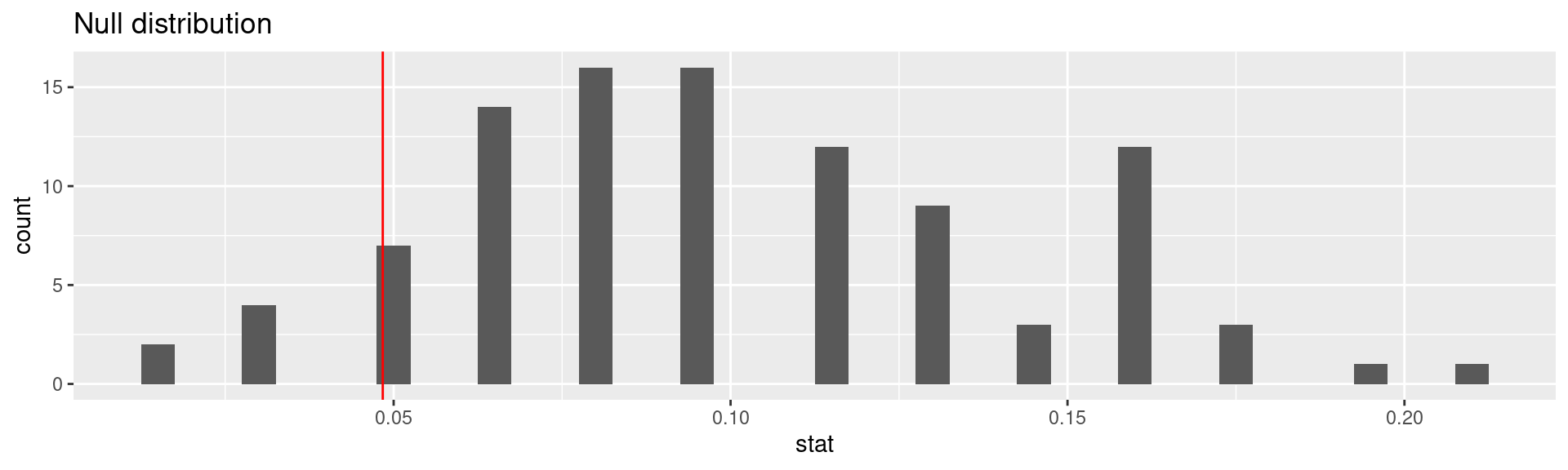
This is the fraction of simulations where the rate of complications was equal or below \(\frac{3}{62} =\) 0.0483871.
Significance level
A significance level\(\alpha\) is a threshold we make up to make our judgment about the plausibility of the null hypothesis being true given the observed data.
We often use \(\alpha = 0.05 = 5\%\) as the cutoff for whether the p-value is low enough that the data are unlikely to have come from the null model.
If p-value < \(\alpha\), reject \(H_0\) in favor of \(H_A\): The data provide convincing evidence for the alternative hypothesis.
If p-value > \(\alpha\), fail to reject \(H_0\) in favor of \(H_A\): The data do not provide convincing evidence for the alternative hypothesis.
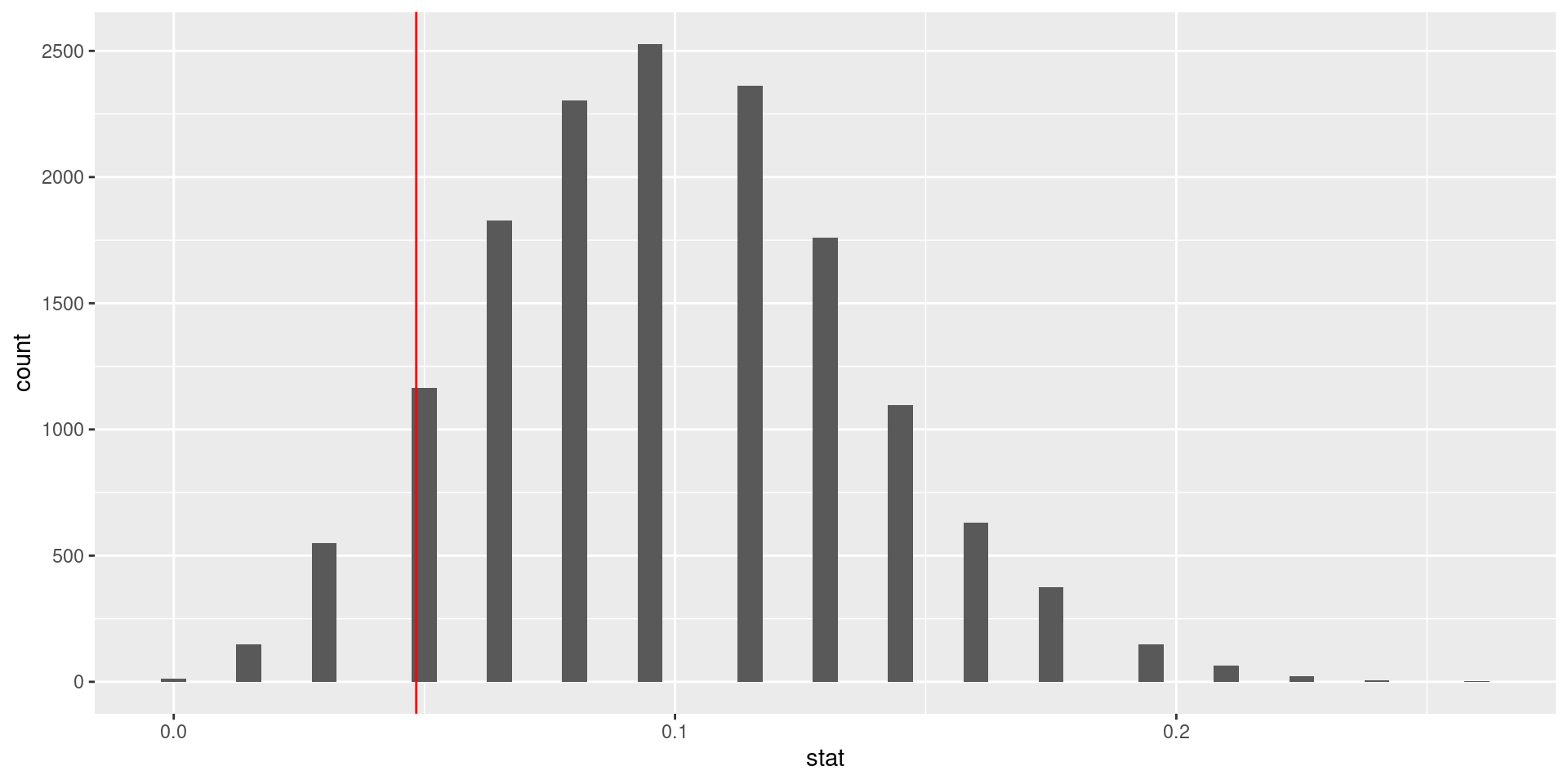
What is the conclusion of the hypothesis test?
Since the p-value is greater than the significance level, we fail to reject the null hypothesis. These data do not provide convincing evidence that this consultant incurs a lower complication rate than the 10% overall US complication rate.
100 simulations is not sufficient
We simulate 15,000 times to get an accurate distribution.
null_dist <- organ_donor |>specify(response = outcome, success ="complication") |>hypothesize(null ="point", p =c("complication"=0.10, "no complication"=0.90)) |>generate(reps =15000, type ="simulate") |>calculate(stat ="prop")ggplot(data = null_dist, mapping =aes(x = stat)) +geom_histogram(binwidth =0.005) +geom_vline(xintercept =3/62, color ="red")
What do the p-values for coefficients mean? What is the null hypothesis?
Null-Hypothesis: No relationship predictor and response. Coefficient could be zero.
Alternative Hypothesis: The coefficient is away from zero.
Small p-value: evidence for rejecting the hypothesis that there is no effect.
Technically: The statistic (which we do not treat now) is sufficiently away from zero.
xkcd on p-values
Significance levels are fairly arbitrary. Sometimes they are used (wrongly) as definitive judgments
They can even be used to do p-hacking: Searching for “significant” effects in observational data
In parts of science it has become a “gamed” performance metric.
The p-value says nothing about effect size!
p-value misinterpretation
p-values do not measure1
the probability that the studied hypothesis is true
the probability that the data were produced by random chance alone
the size of an effect
the importance of a result” or “evidence regarding a model or hypothesis” (it is only against the null hypothesis).
Correct explanation:
The p-value is the probability of obtaining test results at least as extreme as the result actually observed, under the assumption that the null hypothesis is correct.
p-values and significance tests, when properly applied and interpreted, increase the rigor of the conclusions drawn from data.2
Classification: Compare logistic regression and decision tree
Purpose of the section
Go again through the modeling workflow (with tidymodels) and see that large parts are identical
Look again at the coefficients of a logistic regression model
Learn the basic idea of a decision tree (you will not learn the details here)
Do the classification with both models and compare the confusion matrices
Specify recipe and models
For both logistic regression and decision tree:
library(tidymodels)library(palmerpenguins)peng_recipe <-recipe(sex ~ ., data = penguins) |>step_rm(year)
We specify a recipe to predict sex with all available variables in penguins
Typically, more pre-processing steps are specified here, but we are mostly fine
Logistic Regression
peng_logreg <-logistic_reg() |>set_engine("glm")
peng_logreg specifies to fit with glm (generalized linear model from base R)
What do the categorical predictors tell us? Which are signigficant?
What do the numerical predictors tell us? Which are signigficant?
Why is the coefficient for body_mass_g so small, but highly significant?
Categorical predictors: We have 3 species, 3 island. So, we see 4 new variables, 2 for species and 2 for island (the third is the reference category). Species are significant (p < 0.05), but islands not.
Numerical predictors:flipper_length_mm is insignificant, though its coefficient is larger than for body_mass_g. Reason: values of body_mass_g are larger than those of flipper_length_mm. Body mass differs by much more grams than flipper length differs by millimeters.
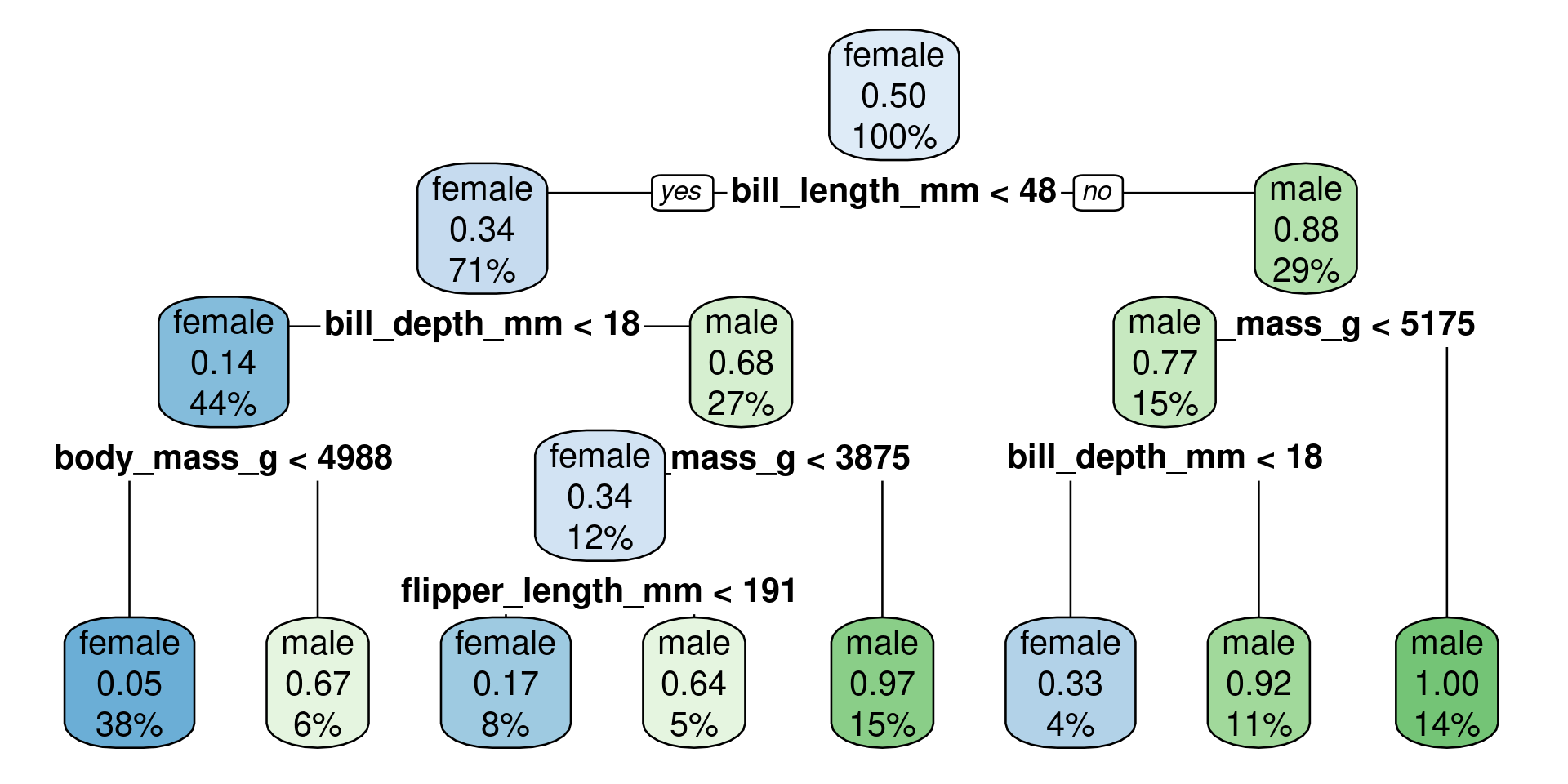
The first three rules would predict female for all observations
The last five rules would predict male for all observations
The order of male and female is because sex is a factor with the first level female and the second level male. The probabilities in front of the rule-text are for the second level: male.
Truth
Prediction female male
female 43 12
male 5 39
The logistic regression has more correct predictions.
Warning: The function conf_mat (from yardstick of tidymodels) shows the transposed confusion matrix compared with Wikipedia:Confusion Matrix. In conf_mat, the true conditions are in columns. The wikipedia convention is that columns are the predicted conditions.
What is a model? Terminological confusion…
“Model” in Statistical Learning
We already had the difference between variable-based and agent-based models in earlier lectures.
But even in the variable-based model setting of statistical learning, the term model can be more or less abstract:
Very general: \(Y = f(X_1, \dots, X_m) + \varepsilon\) where \(Y\) is the response variable and \(X_i\) are features which we put in our model: the abstract and unknown function \(f\). \(\varepsilon\) is the error which can never explain and which we also usually do not know.
More specific: The model\(f\) could already be of a specific type, like linear regression, logistic regression, or a decision tree or other functional forms. As this need not be the real function we may call it assumed model\(\hat f\) For example a linear model \(\hat f(X_1, \dots, X_m) = \beta_0 + \beta_1 X_1 + \dots + \beta_m X_m + \varepsilon\). Now, the model has specified parameters which values are unknown.
More specific: Fitted model
Fitted model: When we have a data set with values for \(Y, X_1, \dots, X_m\) we can fit values for the parameters \(\hat\beta_0, \dots, \hat\beta_m\) to the data. This is the fitted model\(\hat f\). This is called parameter estimation: We estimate \(\hat\beta_0, \dots, \hat\beta_m\) with the hope that they match the real values \(\beta_0, \dots, \beta_m\) and that the linear model \(\hat f\) matches the real function \(f\).
Now we could specify further to fit a specific parameterized model with a specific algorithm, and a specific set of hyperparameters, and maybe more …
Sometimes model means only a certain aspect of all these, for example the formula like sex ~ bill_length_mm + bill_depth_mm + flipper_length_mm + body_mass_g + species + island
Take away: “Model” can mean things with very different granularity. That is OK because they are all related and all fit the definition of being a simplified representation of reality.
Be prepared to specify what you mean when you are talking about a model.
Regression: Compare linear regression and decision tree
Purpose of the section
Go again through the modeling workflow (with tidymodels) and see that large parts are identical
Look again at the coefficients of a linear model
See how the decision tree looks like for a regression problem
Compare the two most common performance measures for regression models: Root Mean Squared Error (RMSE) and R-squared
Specify recipe and models
We specify a recipe to predict body_mass_g with all available variables in penguins and put it in a workflow
Typically, more pre-processing steps are specified here, but we are mostly fine
peng_recipe2 <-recipe(body_mass_g ~ ., data = penguins) |>step_rm(year)peng_workflow2 <-workflow() |>add_recipe(peng_recipe2)
We can re-use the split and the training and test set.
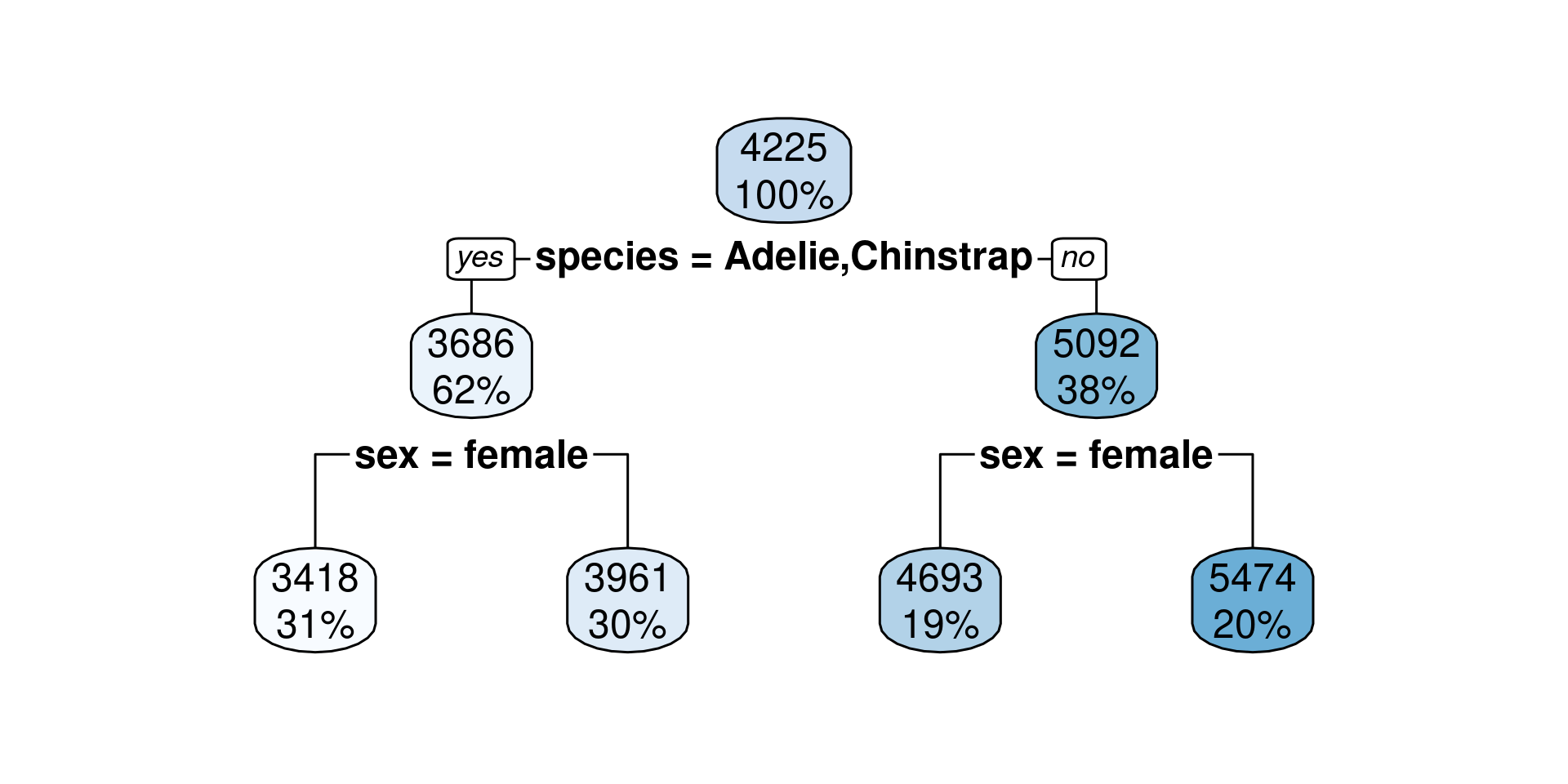
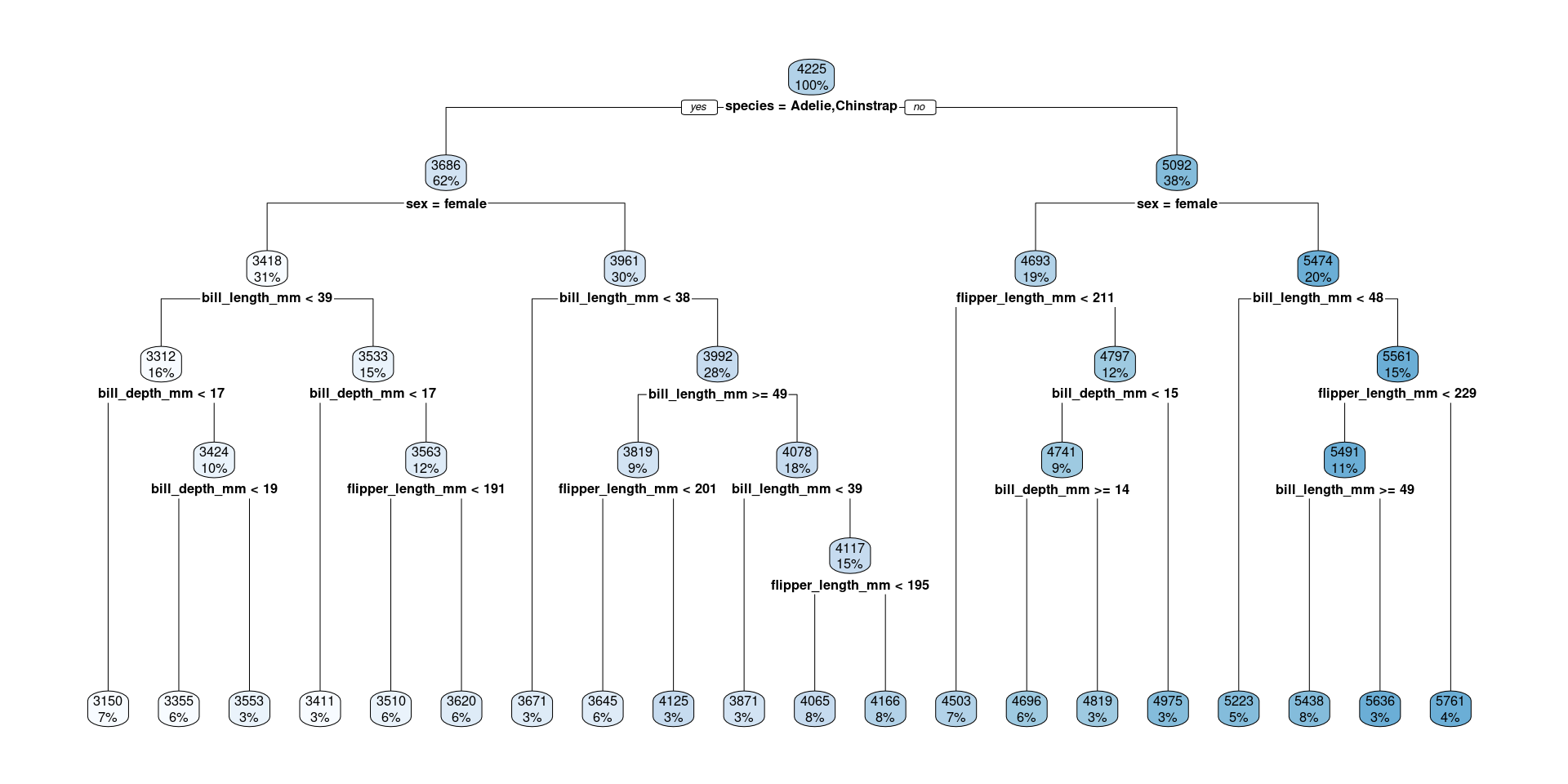
..y
3418 when species is Adelie or Chinstrap & sex is female
3961 when species is Adelie or Chinstrap & sex is male
4693 when species is Gentoo & sex is female
5474 when species is Gentoo & sex is male
For each terminal node a certain value is predicted (the mean of the remaining penguins)
peng_regtree_pred <-predict(peng_regtree_fit, peng_test) |>bind_cols(peng_test)peng_regtree_pred |>select(.pred, body_mass_g, species, sex) |>slice(10*(1:10)) # show same selected rows
# A tibble: 10 × 4
.pred body_mass_g species sex
<dbl> <int> <fct> <fct>
1 3418 3700 Adelie female
2 3961. 3950 Adelie male
3 3961. 3800 Adelie male
4 3961. 3875 Adelie male
5 3418 3400 Adelie female
6 4693. 4150 Gentoo female
7 4693. 4200 Gentoo female
8 4693. 4850 Gentoo female
9 3961. 4150 Chinstrap male
10 3961. 4050 Chinstrap male
Regression Performance Evaluation
R-squared: Percentage of variability in body_mass_g explained by the model
Linear Regression
rsq(peng_linreg_pred, truth = body_mass_g, estimate = .pred)
# A tibble: 1 × 3
.metric .estimator .estimate
<chr> <chr> <dbl>
1 rsq standard 0.861
Decision Tree
rsq(peng_regtree_pred, truth = body_mass_g, estimate = .pred)
# A tibble: 1 × 3
.metric .estimator .estimate
<chr> <chr> <dbl>
1 rsq standard 0.806
Root Mean Squared Error (RMSE):\(\text{RMSE} = \sqrt{\frac{1}{n}\sum_{i = 1}^n (y_i - \hat{y}_i)^2}\)
where \(\hat{y}_i\) is the predicted value and \(y_i\) the true value. (The name RMSE is descriptive.)
rmse(peng_linreg_pred, truth = body_mass_g, estimate = .pred)
# A tibble: 1 × 3
.metric .estimator .estimate
<chr> <chr> <dbl>
1 rmse standard 290.
rmse(peng_regtree_pred, truth = body_mass_g, estimate = .pred)
# A tibble: 1 × 3
.metric .estimator .estimate
<chr> <chr> <dbl>
1 rmse standard 337.
Which model is better in prediction? Linear regression. The R-squared is higher.
What RMSE is better?
Lower. The lower the error, the better the model’s prediction.
It is the other way round than R-squared! Do not confuse them.
Notes:
The common method to fit a linear model is the ordinary least squares (OLS) method
That means the fitted parameters should deliver the lowest possible sum of squared errors (SSE) between predicted and observed values.
Minimizing the sum of squared errors (SSE) is identical to minimizing the mean of squared errors (MSE) because it only adds the factor \(1/n\).
Minimizing the mean of squared errors (MSE) is identical to minimizing the root mean of squared errors (RMSE) because the square root is strictly monotone function.
Conclusion: RMSE can be seen as a definition of the OLS optimization goal.
Interpreting RMSE
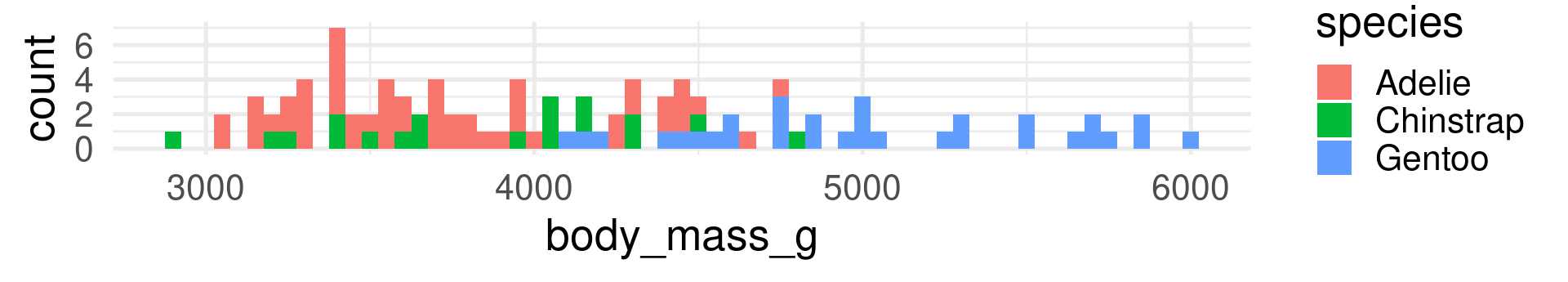
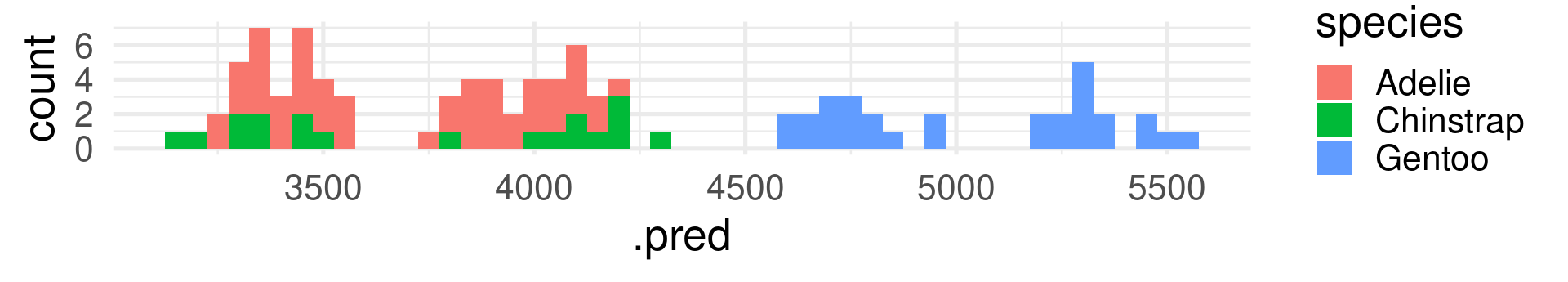
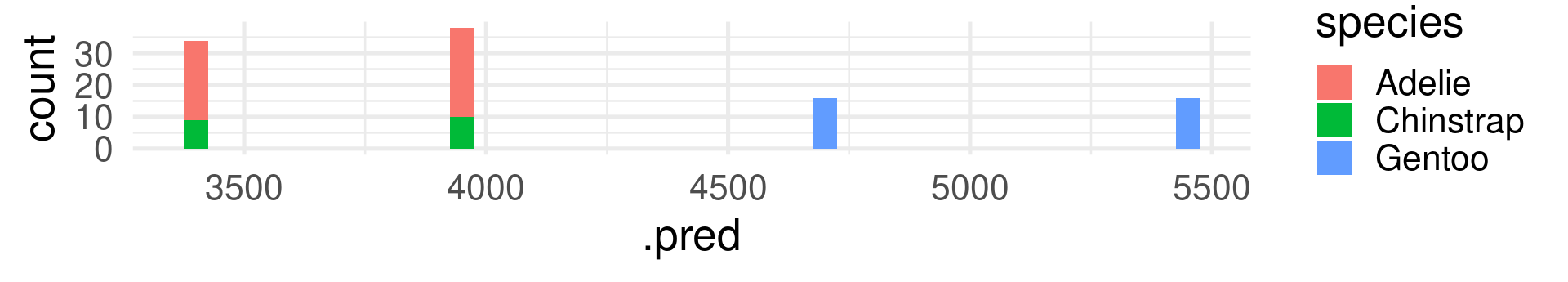
In contrast to R-squared, RMSE can only be interpreted with knowledge about the range and of the response variable! It also has the same unit (grams for body_mass_g).
peng_test |>ggplot(aes(x=body_mass_g, fill = species)) +geom_histogram(binwidth =50) +theme_minimal(base_size =20)
peng_linreg_pred |>ggplot(aes(x=.pred, fill = species)) +geom_histogram(binwidth =50) +theme_minimal(base_size =20)
peng_regtree_pred |>ggplot(aes(x=.pred, fill = species)) +geom_histogram(binwidth =50) +theme_minimal(base_size =20)
The RMSE shows many grams predicted values deviate from the true value on average. (Taking the squaring of differences and root of the average into account.)
# A tibble: 2 × 3
.metric .estimator .estimate
<chr> <chr> <dbl>
1 rmse standard 309.
2 rsq standard 0.857
Where is the prediction better? (Lower RMSE, higher R-squared)
Performance is better for training data (compare values to testing data of the same model). Why? It was used to fit. The model is optimized to predict the training data.
Make a deeper tree
In decision_tree() we can set the maximal depth of the tree to 30.
The trees we had before were also automatically pruned by sensible defaults.
By setting the cost complexity parameter to -1 we avoid pruning.1
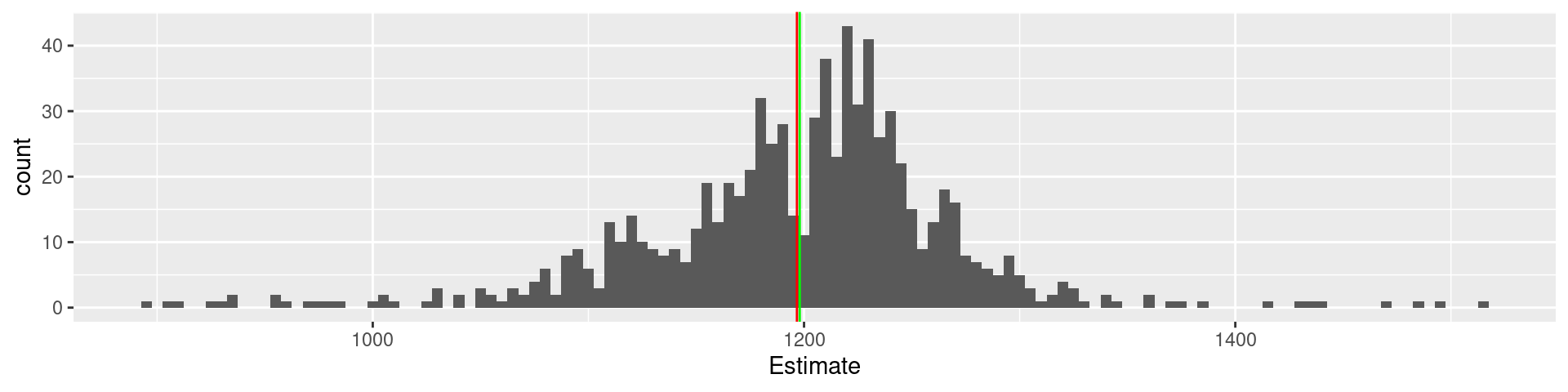
Variance <-var(galton$Estimate)*(nrow(galton)-1)/nrow(galton)# Note, we had to correct for the divisor (n-1) in the classical statistical definition# to get the sample variance instead of the estimate for the population varianceVariance
Variance is a measure for the diversity of estimates around the mean
The mathematical relation \[\text{MSE} = \text{Bias-squared} + \text{Variance}\] can be formulated as
Collective error = Individual error - Diversity
Interpretation: The higher the diversity the lower the collective error!
Why is this message a bit suggestive?
The mathematical relation \[\text{MSE} = \text{Bias-squared} + \text{Variance}\] can be formulated as
Collective error = Individual error - Diversity
Interpretation: The higher the diversity the lower the collective error!
\(\text{MSE}\) and \(\text{Variance}\) are not independent!
Activities to increase diversity (Variance) typically also increase the average individual error (MSE).
For example, if we just add more random estimates with same mean but wild variance to our sample we increase both and do not gain any decrease of the collective error.
Accuracy for numerical estimate
For binary classifiers accuracy has a simple definition: Fraction of correct classifications.
It can be further informed by other more specific measures taken from the confusion matrix (sensitivity, specificity)
How about numerical estimators?
For example outcomes of estimation games, or linear regression models.
Accuracy is for example measured by (R)MSE
\(\text{MSE} = \text{Bias-squared} + \text{Variance}\) shows us that we can make a bias-variance decomposition
That means some part of the error is a systematic (the bias) and another part due to random variation (the variance).
Learn more about the bias-variance tradeoff in statistical learning independently! It is an important concept to understand predictive models.
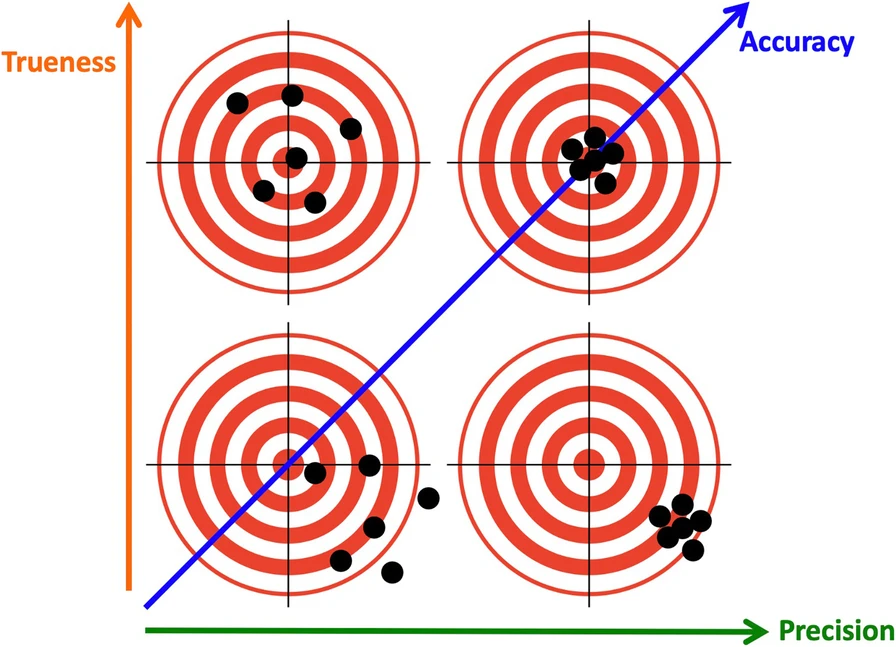
2-d Accuracy: Trueness and Precision
According to ISO 5725-1 Standard: Accuracy (trueness and precision) of measurement methods and results - Part 1: General principles and definitions. there are two dimension of accuracy of numerical measurement.
What is a wise crowd?
Assume the dots are estimates. Which is a wise crowd?
Of course, high trueness and high precision! But, …
Focusing on the crowd being wise instead of its individuals: High trueness, low precision.